DNA Technology
All of the genetic information or DNA in a cell is its genome. Huge doesn't even begin to describe the size of the human genome. It's so big that it can be overwhelming, even to computers. We are usually only interested in studying one or a few genes, not the entire genome. For example, maybe we just want to study a single gene that determines eye color. It's like a needle in the haystack. We just want the needle. To study a single gene, we can create recombinant DNA in the lab. This allows us to study the effects of a gene in another organism. Don't worry; it's not as intimidating as it sounds.
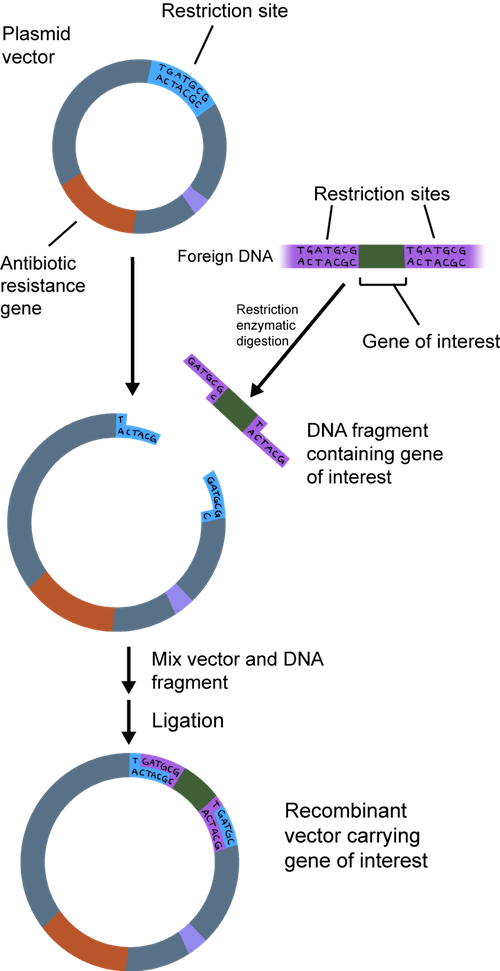
First, the gene needs to hitch a ride to get where it needs to go. You would fly on a plane from the Unites States to get to Europe, right? Well, a single gene cannot enter a completely different organism on its own, either. It needs a souped-up vehicle to get there. What is a gene's vehicle of choice? A loaded bacterial plasmid, of course.
Bacteria have a large, circular chromosome and several smaller plasmids , which don't have many genes. Plasmids are very independent; they don't rely on the chromosome to replicate.
In a nutshell, we can take the gene we are studying and put it into a plasmid. When it is used to make recombinant DNA, the plasmid is called a cloning vector. It is called recombinant because it contains a combination of DNA. The plasmid has all of the bells and whistles to make it easy to insert DNA.
We then put the recombinant DNA into bacteria, which reproduce quickly. The plasmid is replicated so each new bacterium will have the plasmid. Lots of plasmids are made and can be used to make protein encoded by the gene, gene sequencing, or other applications. Now that we've drawn the big picture, it's time jump into the nitty gritty. How, exactly, do scientists make recombinant DNA?
In order to clone a gene into a plasmid, both need to be cut. This is where the DNA scissors, a.k.a. restriction enzymes, come in. These enzymes don't exist just for scientists' cloning pleasure, although that's what they think. Restriction enzymes actually hang around in bacteria to cut up foreign DNA into pieces as a protection mechanism.
The beauty of restriction enzymes is that they cut at very precise sequences of DNA called restriction sites. The fragments that result are called, wait for it…restriction fragments. Why don't restriction enzymes chop up a bacterium's own DNA. It's because their DNA contains methyl groups (–CH3 ) that have been added to adenine and cytosine bases at the sequences recognized by the enzymes. Smart little buggers.
Both the plasmid and the piece of DNA to be cloned into the plasmid are cut by the same restriction enzyme. This generates fragments that have sticky ends. They're not sticky to the touch. They're way too small to be touched. One strand is not base-paired with another strand, leaving a bit of an overhang.
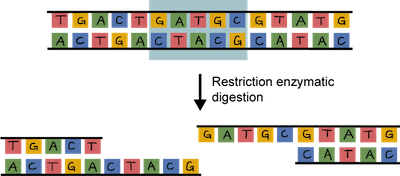
The overhang of the sticky ends of the newly cut plasmid and gene of interest can be glued together by an enzyme called DNA ligase. Ligase acts as a molecular glue and creates complimentary base pairing between the sticky ends.
The recombinant DNA or plasmid can be introduced into bacteria such as E. coli and allowed to replicate. A technique called electroporation that creates temporary pores in the cell membrane is commonly used. An electrical zap is briefly applied to the bacteria creating holes through which the recombinant DNA can enter. Sweet.
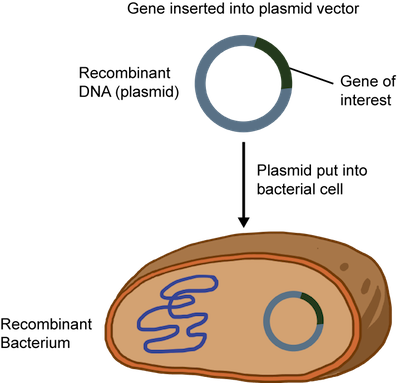
This all sounds great, but how can we be sure that all of the bacteria have the recombinant DNA? The plasmid is usually engineered to contain an antibiotic resistance gene. Remember when we said it had all the bells and whistles? When an antibiotic is added to the medium that the bacteria grows in, only the plasmid containing the resistance gene to that antibiotic will survive. At this point, the plasmid DNA can be isolated and purified from the bacteria.
It's always good to get confirmation that what we have is what we think we have. The quick and dirty check we usually begin with is to cut the purified recombinant DNA with restriction enzymes and check their sizes using electrophoresis.
Cutting the recombinant DNA with the same restriction enzymes we used to make the expression vector in the first place should give us the pieces we started with. We can get a map of the plasmid we used with the restriction sites found in the plasmid. We also know the sequence of the DNA we are inserting. Using these tools, we can determine which restriction enzymes to use and how many and what size fragments should be produced.
It would be awesome if we could actually see the DNA, but we can't. Bummer. Something has to be used to visualize the fragments and to determine their size. Next batter up: electrophoresis.
In DNA electrophoresis, a chemical called agarose is used to form a gel. We can apply a current to the gel and place the negative electrode near the top of the gel where the DNA is loaded and the positive electrode near the bottom of the gel. You know what they say; opposites attract. The negative charges on the phosphate groups of the DNA backbone travel down the gel towards the positive electrode.

Image from here.
The smaller fragments are the speediest while the larger fragments take their good old time traveling through the agarose. A dye that interacts with the DNA, ethidium bromide, is added. Under ultraviolet light, ethidium bromide makes the DNA take on a pink appearance. We can also run DNA of known size on the gel, so the size of the sample fragments can be determined. This method does not provide any information on whether the sequence is the desired one or not. It just helps us determine the size of the fragment.
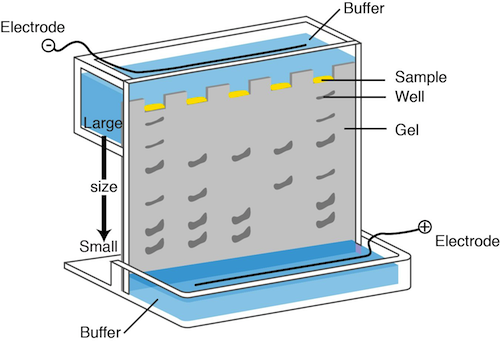
Image from here.
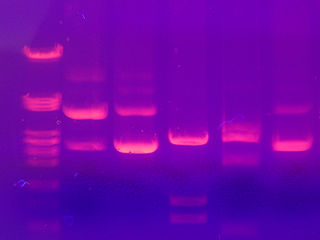
DNA fragments separated by electrophoresis. Image from here.
DNA sequencing can be used to determine the actual sequence of A's, G's, C's and T's. DNA sequencing has come a long way, baby, and it is now quite simple to obtain the DNA sequence of your beloved recombinant DNA. Take a look at the evolution of DNA sequencing.
The original sequencing method was dideoxy chain termination. It sounds pretty scary, but the principle is straightforward. Double stranded DNA is heated and the strands are separated or denatured. A short sequence of nucleotides called a primer is used to bind to a known sequence near the 3' end of the DNA. DNA polymerase, the four dNTPs (dATP, dCTP, dGTP, and dTTP), and the primers are added to the tube. An altered dNTP called a dideoxynucleotide is tattooed with a fluorescent tag and added to the tube as well. The thing is, these dideoxynucleotides, when incorporated, are sneaky and stop the DNA chain from growing. They are incorporated randomly. The result? Lots and lots of DNA fragments of various sizes, each with a fluorescent tag on its 3' end.
What should we do with all of these fragments? They can be separated based on their size through a gel matrix. We use a gel called polyacrylamide that has tiny pores. The smallest fragments move the quickest. The largest move the slowest.
A machine that detects fluorescence travels up the gel reading the dideoxynucleotides, and voilà, there's the sequence. It's actually the sequence of the complementary strand, but the original sequence can be determined from this info.
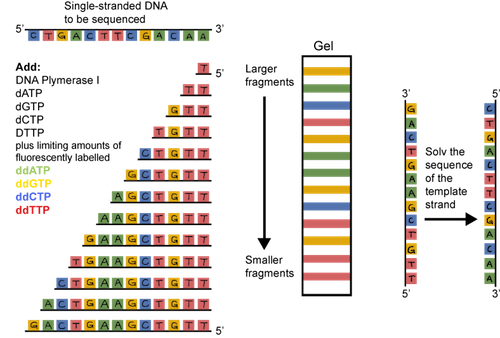
Sounds pretty tedious, doesn't it? It is. Thankfully, DNA sequencing has evolved. Now, it's easier and cheaper than ever to have DNA sequenced. You can sleep more easily at night.
Chain termination has given rise to dangling a single-strand of DNA on a surface. Think of it as the worm when you go fishing. The dangling piece of DNA looks yummy to its complementary bases. They are added and can be detected by a machine. Much simpler, eh? The sequence can literally be read from the print out of data.
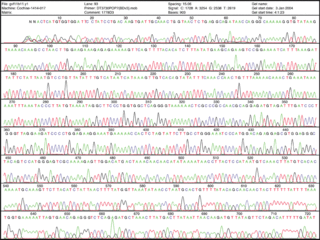
See enlarged image here.
Now that's a good lookin' sequence!
First, the gene needs to hitch a ride to get where it needs to go. You would fly on a plane from the Unites States to get to Europe, right? Well, a single gene cannot enter a completely different organism on its own, either. It needs a souped-up vehicle to get there. What is a gene's vehicle of choice? A loaded bacterial plasmid, of course.
Bacteria have a large, circular chromosome and several smaller plasmids , which don't have many genes. Plasmids are very independent; they don't rely on the chromosome to replicate.
In a nutshell, we can take the gene we are studying and put it into a plasmid. When it is used to make recombinant DNA, the plasmid is called a cloning vector. It is called recombinant because it contains a combination of DNA. The plasmid has all of the bells and whistles to make it easy to insert DNA.
We then put the recombinant DNA into bacteria, which reproduce quickly. The plasmid is replicated so each new bacterium will have the plasmid. Lots of plasmids are made and can be used to make protein encoded by the gene, gene sequencing, or other applications. Now that we've drawn the big picture, it's time jump into the nitty gritty. How, exactly, do scientists make recombinant DNA?
In order to clone a gene into a plasmid, both need to be cut. This is where the DNA scissors, a.k.a. restriction enzymes, come in. These enzymes don't exist just for scientists' cloning pleasure, although that's what they think. Restriction enzymes actually hang around in bacteria to cut up foreign DNA into pieces as a protection mechanism.
The beauty of restriction enzymes is that they cut at very precise sequences of DNA called restriction sites. The fragments that result are called, wait for it…restriction fragments. Why don't restriction enzymes chop up a bacterium's own DNA. It's because their DNA contains methyl groups (–CH3 ) that have been added to adenine and cytosine bases at the sequences recognized by the enzymes. Smart little buggers.
Both the plasmid and the piece of DNA to be cloned into the plasmid are cut by the same restriction enzyme. This generates fragments that have sticky ends. They're not sticky to the touch. They're way too small to be touched. One strand is not base-paired with another strand, leaving a bit of an overhang.
The overhang of the sticky ends of the newly cut plasmid and gene of interest can be glued together by an enzyme called DNA ligase. Ligase acts as a molecular glue and creates complimentary base pairing between the sticky ends.
The recombinant DNA or plasmid can be introduced into bacteria such as E. coli and allowed to replicate. A technique called electroporation that creates temporary pores in the cell membrane is commonly used. An electrical zap is briefly applied to the bacteria creating holes through which the recombinant DNA can enter. Sweet.
This all sounds great, but how can we be sure that all of the bacteria have the recombinant DNA? The plasmid is usually engineered to contain an antibiotic resistance gene. Remember when we said it had all the bells and whistles? When an antibiotic is added to the medium that the bacteria grows in, only the plasmid containing the resistance gene to that antibiotic will survive. At this point, the plasmid DNA can be isolated and purified from the bacteria.
It's always good to get confirmation that what we have is what we think we have. The quick and dirty check we usually begin with is to cut the purified recombinant DNA with restriction enzymes and check their sizes using electrophoresis.
Cutting the recombinant DNA with the same restriction enzymes we used to make the expression vector in the first place should give us the pieces we started with. We can get a map of the plasmid we used with the restriction sites found in the plasmid. We also know the sequence of the DNA we are inserting. Using these tools, we can determine which restriction enzymes to use and how many and what size fragments should be produced.
It would be awesome if we could actually see the DNA, but we can't. Bummer. Something has to be used to visualize the fragments and to determine their size. Next batter up: electrophoresis.
In DNA electrophoresis, a chemical called agarose is used to form a gel. We can apply a current to the gel and place the negative electrode near the top of the gel where the DNA is loaded and the positive electrode near the bottom of the gel. You know what they say; opposites attract. The negative charges on the phosphate groups of the DNA backbone travel down the gel towards the positive electrode.
Image from here.
The smaller fragments are the speediest while the larger fragments take their good old time traveling through the agarose. A dye that interacts with the DNA, ethidium bromide, is added. Under ultraviolet light, ethidium bromide makes the DNA take on a pink appearance. We can also run DNA of known size on the gel, so the size of the sample fragments can be determined. This method does not provide any information on whether the sequence is the desired one or not. It just helps us determine the size of the fragment.
Image from here.
DNA fragments separated by electrophoresis. Image from here.
DNA sequencing can be used to determine the actual sequence of A's, G's, C's and T's. DNA sequencing has come a long way, baby, and it is now quite simple to obtain the DNA sequence of your beloved recombinant DNA. Take a look at the evolution of DNA sequencing.
The original sequencing method was dideoxy chain termination. It sounds pretty scary, but the principle is straightforward. Double stranded DNA is heated and the strands are separated or denatured. A short sequence of nucleotides called a primer is used to bind to a known sequence near the 3' end of the DNA. DNA polymerase, the four dNTPs (dATP, dCTP, dGTP, and dTTP), and the primers are added to the tube. An altered dNTP called a dideoxynucleotide is tattooed with a fluorescent tag and added to the tube as well. The thing is, these dideoxynucleotides, when incorporated, are sneaky and stop the DNA chain from growing. They are incorporated randomly. The result? Lots and lots of DNA fragments of various sizes, each with a fluorescent tag on its 3' end.
What should we do with all of these fragments? They can be separated based on their size through a gel matrix. We use a gel called polyacrylamide that has tiny pores. The smallest fragments move the quickest. The largest move the slowest.
A machine that detects fluorescence travels up the gel reading the dideoxynucleotides, and voilà, there's the sequence. It's actually the sequence of the complementary strand, but the original sequence can be determined from this info.
Sounds pretty tedious, doesn't it? It is. Thankfully, DNA sequencing has evolved. Now, it's easier and cheaper than ever to have DNA sequenced. You can sleep more easily at night.
Chain termination has given rise to dangling a single-strand of DNA on a surface. Think of it as the worm when you go fishing. The dangling piece of DNA looks yummy to its complementary bases. They are added and can be detected by a machine. Much simpler, eh? The sequence can literally be read from the print out of data.
See enlarged image here.
Now that's a good lookin' sequence!